
Prof.Dr. Haydar BAĞIŞ
ADYÜ Tıp Fak. Dekan V.
Tıbbi Genetik ABD Başkanı.
14.05.2025
Marfan Sendromu ve Ailevi Torasik Aort Anevrizması ve Diseksiyonunda Genetiğin Rolü
Her 5.000 kişiden yaklaşık 1'inde Marfan sendromu vardır, buna tüm ırklardan ve etnik gruplardan erkekler ve kadınlar dahildir. Marfan sendromu olan yaklaşık 4 kişiden 3'ü bunu miras alır, yani genetik mutasyonu bu sendroma sahip bir ebeveynden alırlar. Ancak Marfan sendromu olan bazı kişiler ailelerinde bu sendroma sahip olan ilk kişilerdir; bu olduğunda buna kendiliğinden mutasyon (de Novo) denir. Marfan sendromu olan bir kişinin her çocuk sahibi olduğunda genetik mutasyonu aktarma olasılığı %50'dir.
Bağ dokusu vücudun her yerinde bulunduğundan, Marfan sendromu vücudun birçok farklı bölümünü de etkileyebilir. Bozukluğun özellikleri en sık kalpte, kan damarlarında, kemiklerde, eklemlerde ve gözlerde bulunur. Bazı Marfan özellikleri -örneğin, aort genişlemesi (kanı kalpten vücudun geri kalanına taşıyan ana kan damarının genişlemesi)- yaşamı tehdit edici olabilir. Akciğerler, cilt ve sinir sistemi de etkilenebilir. Marfan sendromu zekayı etkilemez.
Marfan sendromunun belirtileri ve semptomları, aynı aileden gelen üyeler arasında bile büyük ölçüde farklılık gösterebilir, çünkü bu bozukluk vücudun birçok farklı bölgesini etkileyebilir. Bazı kişiler yalnızca hafif etkiler yaşarken, diğerleri yaşamı tehdit eden komplikasyonlar geliştirir.
Marfan sendromunun özellikleri şunları içerebilir:
- Uzun ve ince yapılı
- Orantısız derecede uzun kollar, bacaklar ve parmaklar
- Dışarı doğru çıkıntı yapan (pectus carinatum) veya içeriye doğru çöken göğüs kemiği (pectus excavatum)
- Yüksek, kemerli damak ve sıkışık dişler
- Kalp üfürümleri
- Aşırı miyopluk
- Anormal derecede kavisli bir omurga
- Düz taban
Marfan Sendromunda Genetik Testlerin Rolü
FBN1 geninin Marfan sendromuna nedeni olduğu bulunduktan sonra, Marfan sendromunun tanısında genetik testlerin rolünü ve Marfan sendromunun belirli klinik özelliklerinin belirli FBN1 mutasyonlarıyla ilişkilendirilip ilişkilendirilemeyeceğini belirlemek için testler yapıldı. Mutasyon tanımlaması artık Marfan sendromunun tanı kriterlerine dahil edilmiştir ve bir FBN1 gen mutasyonu tanımlanmazsa, Marfan sendromu tanısı sorgulanmalıdır. (“Marfan sendromu için revize edilmiş Gent nosolojisi.” J Med Genet; 47:476-485 doi:10.1136/jmg.2009.072785)
FBN1 geninin patogenetik varyantları
FBN1 geni, kromozom 15'in uzun kolunda yer alır ve 65 kodlama ekzonuna sahiptir. Marfana yatkınlık oluşturan nadir patojenik varyantlar gen boyunca dağılmıştır. Bugüne kadar FBN1'de yaklaşık 2.000 nadir varyant tanımlanmıştır ve bunların çoğu Marfana yatkınlık oluşturur.
Marfan benzeri özelliklere sahip ancak hipertelorizm (gözlerin birbirinden uzak olması) ve bifid uvula gibi bazı ayırt edici bulgulara sahip bir grup bireyde TGFß reseptör genlerindeki (TGFBR1 ve TGFBR2) mutasyonlar belirlendi. Daha sonra ACTA2, MYH11, MYLK, PRKG1, SMAD3 ve TGFB2'deki mutasyonların belirlenmesi, diğer genlerdeki mutasyonlar nedeniyle aort hastalığı olan birey gruplarını ayırdı. Önemlisi, bu diğer genlerde mutasyonu olan bireylerin aort hastalıklarının genellikle Marfan sendromlu hastalardan farklı şekilde yönetilmesi gerekir. Ek olarak, bu diğer genlerden bazıları Marfan sendromlu hastalarda olmayan diğer atardamarlarla ilgili sorunlar için risk oluşturur.
FBN1 geninin genetik analizi şu anda yaklaşık 3 hafta sürüyor. Marfan sendromlu çoğu kişide genin nüklotidlerinden yalnızca bir veya birkaçını değiştiren değişiklikler vardır (mutasyon) ve bunlar kolayca belirlenir. Ancak bazılarında genin birçok nükleotidini silen (büyük delesyon) veya çoğaltan (dublikasyon) mutasyonlar vardır. Bu mutasyonlar mevcut dizileme stratejileri tarafından tespit edilmez ve ek testler yapılması gerekir, bu da 2-3 hafta daha ekleyebilir. Genetik test maliyetleri hızla düşmektedir.
Ailedeki bir bireyde mutasyon tanımlandığında, risk altında olan diğer tüm aile üyeleri (ebeveynler, kardeşler, çocuklar) her biri ucuza test edilebilir, çünkü etkilenen her aile üyesinde gende aynı değişiklik veya mutasyon olacaktır. Risk altında olan aile üyelerinin genetik testi, mutasyona sahip olan aile üyelerini mutasyona sahip olmayanlardan ayırt edebilir, bu da aile için güçlü bir bilgidir. Mutasyona sahip aile üyeleri, aort diseksiyonlarını önlemek için aort hastalığı açısından taranabilir ve takip edilebilir.
Mutasyona sahip olmayan aile üyelerinin aort hastalığı açısından taranmasına gerek yoktur. Gerçekten de, aort hastalığını taramak için görüntüleme maliyeti, genetik test maliyetinden çok daha fazladır. Şu anda ekokardiyografi çalışma başına 2.000 dolara mal oluyor ve bilgisayarlı tomografik anjiyografi (CTA) veya manyetik rezonans anjiyografi (MRA) 5.000 dolar veya daha fazlaya mal olabilir.
Genetik testin birey için ne gibi bir faydası var? Marfan sendromunun açıkça kesin bir tanısı karşısında (örneğin tanı ve lens subluksasyonu ile uyumlu iskelet bulguları), test bir şey katıyor mu? Bazı durumlarda, cevap evettir. Örneğin, fibrolin proteininin 233. pozisyonundaki bir arginini bir sisteinle (Arg233Cys) değiştiren bir mutasyon, lens subluksasyonu ve belirgin iskelet değişiklikleri olan kişilerde görülür, ancak aortları genişleme eğiliminde değildir veya çok uzun bir süre boyunca aşırı yavaş genişler.
"Genetik testler, yalnızca aile üyelerini test etmek ve kimin risk altında olduğunu anlamak için kullanılacak bir gen varyantı bulmamıza yardımcı olmakla kalmıyor, aynı zamanda sonuçlar, tespit edilen belirli gene ve muhtemelen varyanta dayalı olarak uzmanlaşmış tıbbi yönetim sağlayarak sağlık hizmetinizi "ayarlamamıza" da yardımcı olabilir."
Torasik aort anevrizması ve diseksiyonu, torasik aortun genişlemesi ve/veya diseksiyonunun varlığına ve Marfan sendromu, Loeys-Dietz sendromu veya vasküler Ehlers-Danlos sendromu gibi diğer bağ dokusu bozukluklarının klinik özelliklerinin yokluğuna dayanarak teşhis edilir. Bu bağ dokusu bozuklukları için ayırıcı tanı ve muhtemelen birden fazla geni tarayan bir genetik test (aort paneli) yaptırmak hakkında doktorunuzla görüşün.
Ek olarak, Marfan sendromu ile Loeys-Dietz sendromu arasındaki ayrım klinik olarak belirgin olmayabilir, ancak genetik testlerle kolayca yapılabilir. Yine de, Marfan sendromu veya Loeys-Dietz sendromu olan bir bireyi doğru şekilde teşhis etmek, aort anevrizmalarının yönetiminde fark yaratır ve diğer atardamarların görüntülenmesi için gereken miktarı belirler. T
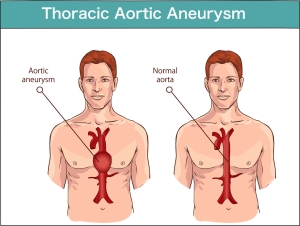
Torasik aort anevrizması, torasik boşlukta veya göğüs bölgesinde aortun (kalpten vücudun geri kalanına kan taşıyan ana kan damarı) genişlemesidir . Bu durum genel nüfusta oldukça yaygındır, ancak vakaların yaklaşık %20'si genetik bir durumdan kaynaklanır.
Erken teşhis, sık görüntüleme ve tedavi kritik öneme sahiptir çünkü hem aort anevrizmaları hem de diseksiyonlar aortun aniden patlaması (yırtılması) ve büyük iç kanamaya neden olma riskini artırır. Aort yırtılmasını önlemek için ameliyat yapılmazsa, bu kan damarı anormallikleri yaşamı tehdit edici olabilir.
- Aort anevrizması. Kalbinizden çıkan kanın basıncı, aort duvarınızın lastikteki zayıf bir nokta gibi dışarı doğru şişmesine neden olabilir. Marfan sendromu olan kişilerde, bunun en çok aort kökünde, yani atardamarın kalbinizden ayrıldığı yerde meydana gelmesi olasıdır.
- Aort diseksiyonu. Aort duvarı katmanlardan oluşur. Diseksiyon, aort duvarının en iç katmanındaki küçük bir yırtık kanın duvarın iç ve dış katmanları arasına sıkışmasına izin verdiğinde meydana gelir. Bu göğüste veya sırtta şiddetli ağrıya neden olabilir. Aort diseksiyonu damarın yapısını zayıflatır ve ölümcül olabilen bir yırtılmaya neden olabilir.
- Kapak malformasyonları. Marfan sendromu olan kişilerde kalp kapakçıklarında zayıf doku olabilir. Bu, kapakçık dokusunun gerilmesine ve anormal kapakçık işlevine neden olabilir. Kalp kapakçıkları düzgün çalışmadığında, kalbiniz genellikle telafi etmek için daha fazla çalışmak zorunda kalır. Bu, sonunda kalp yetmezliğine yol açabilir.
Ailevi Torasik Aort Anevrizması ve Diseksiyonunun Temel Özellikleri Nelerdir?
Aort genişlemesi (dilatasyon/anevrizma) genellikle bu durumun ana özelliğidir. Aort diseksiyonu, aortun iç duvarının aortun iç ve dış duvarları arasında kan akışına izin veren aortun iç duvarının aniden yırtılmasıdır. Bu, göğüs ve/veya sırt ağrısına neden olur ve aort yırtılmasına yol açabilir.
İnsanlar hayatlarının herhangi bir döneminde anevrizma veya aort diseksiyonu geliştirebilirler. Bu durum kalıtsal olabilir ve aynı aile içinde bile bu sorunların ortaya çıkışı ve zamanlaması değişebilir.
Ailevi Torasik Aort Anevrizması ve Diseksiyonunun Nedenleri Nelerdir?
Torasik aort anevrizması ve diseksiyonu olan kişilerin yaklaşık %20'sinde buna genetik yatkınlık vardır, yani ailede görülür. Bu türe ailesel torasik anevrizma ve diseksiyon denir. Birçok kişi torasik aort anevrizması ve diseksiyonuna genetik yatkınlıkları olduğunu bilmez. Torasik aort anevrizması olduğu bilinen kişilerin birinci derece akrabaları (yani ebeveynleri, çocukları, kardeşleri) bu durum için taranmalıdır.
Ailevi torasik aort anevrizması ve diseksiyonu tanısı, ailede anevrizma ve diseksiyon öyküsünün olmasıyla konur.
Aort anevrizmaları ekokardiyografi (ses dalgası resmi), bilgisayarlı tomografi (BT veya CAT taraması), manyetik rezonans görüntüleme (MRI), transözofageal ekokardiyogram (TEE), göğüs röntgeni veya anjiyografi gibi görüntüleme teknikleri kullanılarak teşhis edilir. Aort diseksiyonları bilgisayarlı tomografi (BT veya CAT taraması) veya transözofageal ekokardiyogram (TEE) ile teşhis edilebilir. FTAAD'ye neden olduğu bilinen birçok gen vardır ancak hala keşfedilmesi gereken birçok gen vardır.
Ailevi Torasik Aort Anevrizması ve Diseksiyonu Nasıl Tedavi Edilir?
- Ailevi torasik aort anevrizması ve/veya diseksiyonunun yönetimi, tıbbi genetikçi, kardiyolog ve kardiyovasküler cerrah gibi bu duruma aşina olan multidisipliner bir uzman ekibinin koordineli çalışmasını gerektirir.
- Aort üzerindeki stresi azaltan ilaçlar (ilaçlar) yardımcı olabilir. Beta blokerler kan basıncını düşürmeye ve kalp atışının kuvvetini azaltmaya yardımcı olur. Ayrıca aort genişlemesini önlemeye veya yavaşlatmaya ve aort diseksiyonu riskini azaltmaya yardımcı olabilirler.
- Ailevi torasik aort anevrizması ve diseksiyonu olan kişiler, acil bir durum olmadan önce aort sağlıklarını izlemek ve sorunları belirlemek için rutin testlere tabi tutulmalıdır. Bunlar, doktorların durumu teşhis etmek için kullandıkları görüntüleme testleriyle aynıdır: ekokardiyogram, MRI, BT taraması veya TEE.
- ACTA2 mutasyonu olan bireyler için koroner arter hastalığı ve serebrovasküler hastalık taraması makuldür. TGFBR1, TGFBR2, TGFB2 veya SMAD3 mutasyonu olan bireyler için aort ve dallarının ve serebrovasküler dolaşımın yıllık görüntülenmesi önerilir.
Ameliyat, aort diseksiyonu veya diğer yaşamı tehdit eden bir durumdan önce yapılırsa en etkilidir.
Ameliyat şu durumlarda düşünülür:
- Yükselen aortun genişleme hızı yılda 0,5 cm'ye yaklaşır
- Yükselen aortun çapı 4,2 ila 5,0 cm arasındadır (altta yatan mutasyona veya aile öyküsüne bağlı olarak)
En sık uygulanan iki ameliyat türü şunlardır:
- Aort greft onarımı ve aort mekanik kapakçığı takılmasını içeren Bentall prosedürü. Bu ameliyattan sonra, hastanın kan pıhtılarını önlemek için hayatının geri kalanında kan sulandırıcı ilaç alması gerekir, bu da yaşamı tehdit edici olabilir.
- Aortun hasarlı kısmını değiştiren ancak hastanın kendi aort kapağını koruyan kapak koruyucu cerrahi. Sonrasında kan sulandırıcı ilaç gerektirmeyen bu tür cerrahi, yalnızca hastanın kapağı düzgün çalışıyorsa yapılabilir.
- Ameliyatın türü ne olursa olsun, hastalar tansiyon ilaçlarını almaya devam etmeli ve aortun diğer kısımlarında ek genişlemelere veya yırtıklara karşı koruma sağlamak için yılda en az bir kez aortlarını izlettirmelidirler.
Ailevi Torasik Aort Anevrizması ve Diseksiyonu ve Gebelik:
Aile planlamasıyla ilgili kararlar, olası ebeveynlerden birinin ailevi torasik aort anevrizması gibi genetik bir rahatsızlığı olduğunda zor ve çok duygusal olabilir. Herhangi bir karar vermeden önce, ebeveynler şu anda mevcut olan birçok planlama seçeneğini ve çocuk ve anne için olası riskleri anlamalıdır. Bir genetik danışman sizinle seçenekleri tartışabilir ve öngörü sağlayabilir.
Yükselen aort kökü anevrizması onarımı ve replasmanı:
Yükselen aort kökü anevrizması onarımı ve replasmanı iki şekilde yapılabilir. Kapakçık koruyan aort kökü onarımı (sağ üstteki resim), aortun genişlemiş kısmını greft adı verilen yapay bir tüple değiştirir. Aort kapağı yerinde kalır. Aort kapağı ve aort kökü replasmanında (sağ alttaki resim), kapak ve aortun bir kısmı çıkarılır. Aortun bir kısmı bir greftle değiştirilir. Kapakçığın yerini mekanik veya biyolojik bir kapak alır.
Fiziksel aktivite
Yoğun fiziksel aktivite, etkilenen aortunuzu ve diğer bağ dokularınızı zorlayabilir. Bu nedenle, sizin için güvenli olan egzersizleri ve sporları bulmak için bir fizyoterapistle yakın bir şekilde çalışacaksınız .
Genellikle marfanlı çoğu kişi için düşük ila orta yoğunlukta egzersiz önerilmektedir. Sağlığınız için daha güvenli olabilecek aktiviteler arasında yürüyüş, bisiklet sürme ve yüzme yer almaktadır.
Genel olarak şu sporlardan kaçınmanız gerekebilir:
- Temas sporları
- Yorgunluğa kadar egzersiz
KAYNAKLAR:
https://www.niams.nih.gov/health-topics/marfan-syndrome/more-info/
https://marfan.org/conditions/marfan-syndrome/
https://www.mayoclinic.org/diseases-conditions/marfan-syndrome/symptoms-causes/syc-20350782




